WGS/WTS bei AML und ALL
Das Ungewöhnliche erkennen ohne die diagnostische Genauigkeit zu verlieren - eine prospektive WGS/WTS-Pilotstudie bei akuten Leukämien liefert zusätzliche Informationen für Diagnose, Prognose und Behandlung
Hintergund
Gemäß den Leitlinien der WHO (World Health Organization) und des ELN (European Leukemia Net) basieren die Standarddiagnostik und die Risikostratifizierung für akute Leukämien (AML, akute myeloische Leukämie und ALL, akute lymphatische Leukämie) auf genetischen Informationen, die durch eine Kombination aus Zytogenetik und molekulargenetischen Mutationsprofilen gewonnen werden. Heutzutage können umfassende Techniken, wie WGS und WTS (WGTS, Ganzgenom- und Transkriptomsequenzierung) und die dazugehörigen klinischen Bioinformatik-Workflows die Diagnostik noch verfeinern und individualisieren. In diesem Projekt soll der klinische Nutzen und der Mehrwert von WGTS in einem realen diagnostischen Setting im Vergleich zur genetischen Standarddiagnostik aus Zytogenetik und molekulargenetischer Panel-Sequenzierung evaluiert werden.
Ergebnisse
An der Studie nahmen bisher 113 Patienten teil, 97 mit AML und 16 mit ALL, die nach den WHO-Kriterien auf Grundlage der Zytomorphologie, Immunphänotypisierung, Zyto- und Molekulargenetik diagnostiziert wurden. Für ausgewählte AML-Fälle wurden CD3+ MAC-sortierte Zellen als Keimbahn-Kontrollen mitgeführt.
Alle Patienten wurden auf Strukturvarianten (SV), Kopienzahlvariationen (CNV) und Kopienzahl-neutralen Verlust der Heterozygotie (cnLOH) auf Grundlage von WGS-Daten sowie auf das Vorhandensein von Fusionstranskripten und Expressionsmustern auf Grundlage von WTS-Daten untersucht. Proben ohne eine Keimbahn-Kontrolle (n = 74) wurden auf 121 Gene untersucht, die bei myeloischen/lymphatischen Neoplasien häufig mutiert sind, während bei 39 AML-Fällen mit CD3+ Keimbahn-Kontrollen das gesamte Exom untersucht wurde.
Insgesamt wurden 308/333 (92 %) Genmutationen durch Panel-Sequenzierung und WGS übereinstimmend identifiziert. Aufgrund der höheren Lesetiefe wurden 25/333 (8 %) subklonale Mutationen nur durch gezielte Panel-Sequenzierung nachgewiesen, wobei 23/25 eine Allelfraktion <10 % aufwiesen. Allerdings wurden 40 Genmutationen aufgrund des uneingeschränkten Analysespektrums ausschließlich durch WGS identifiziert. Jeder Patient trug im Durchschnitt 3,3 Genmutationen innerhalb der 121 untersuchten Gene. Bei der Analyse des Exoms erhöhte sich diese Zahl auf 63 Varianten (Mittelwert; Range: 46-116). Bei 52/63 Varianten handelt es sich höchst wahrscheinlich um Keimbahnvarianten, da sie auch in den CD3+ Kontrollproben vorkommen. Die CD3+-Kontrollproben reduzierten die Zahl der somatischen Varianten im Exom damit auf 11 (Mittelwert; Range: 1-23), bei denen es sich meist um patientenspezifische Mutationen handelt, die für personalisierte Therapieansätze in Betracht gezogen werden können.
In Bezug auf Zytogenetik, die für die Risikostratifizierung und die Zuordnung zu genetischen Untergruppen der AML/ALL von Bedeutung ist, identifizierte WTS alle entitätsdefinierenden Fusionen. WGS und die Chromosomen-Banden-Analyse (CBA) zeigten eine sehr hohe Übereinstimmung. Bei 10 Patienten wurden mit WGS subklonale Aberrationen aufgrund der geringen Klongröße übersehen (mediane Klongröße bei IP-FISH-Analyse: 6 %). Allerdings lieferte WGS bei 40 Patienten zusätzliche Informationen, meist SV, CNV und cnLOH von unklarer Signifikanz, aber auch Veränderungen von biologischem Interesse und prognostischer und/oder therapeutischer Bedeutung (Abb. 1):
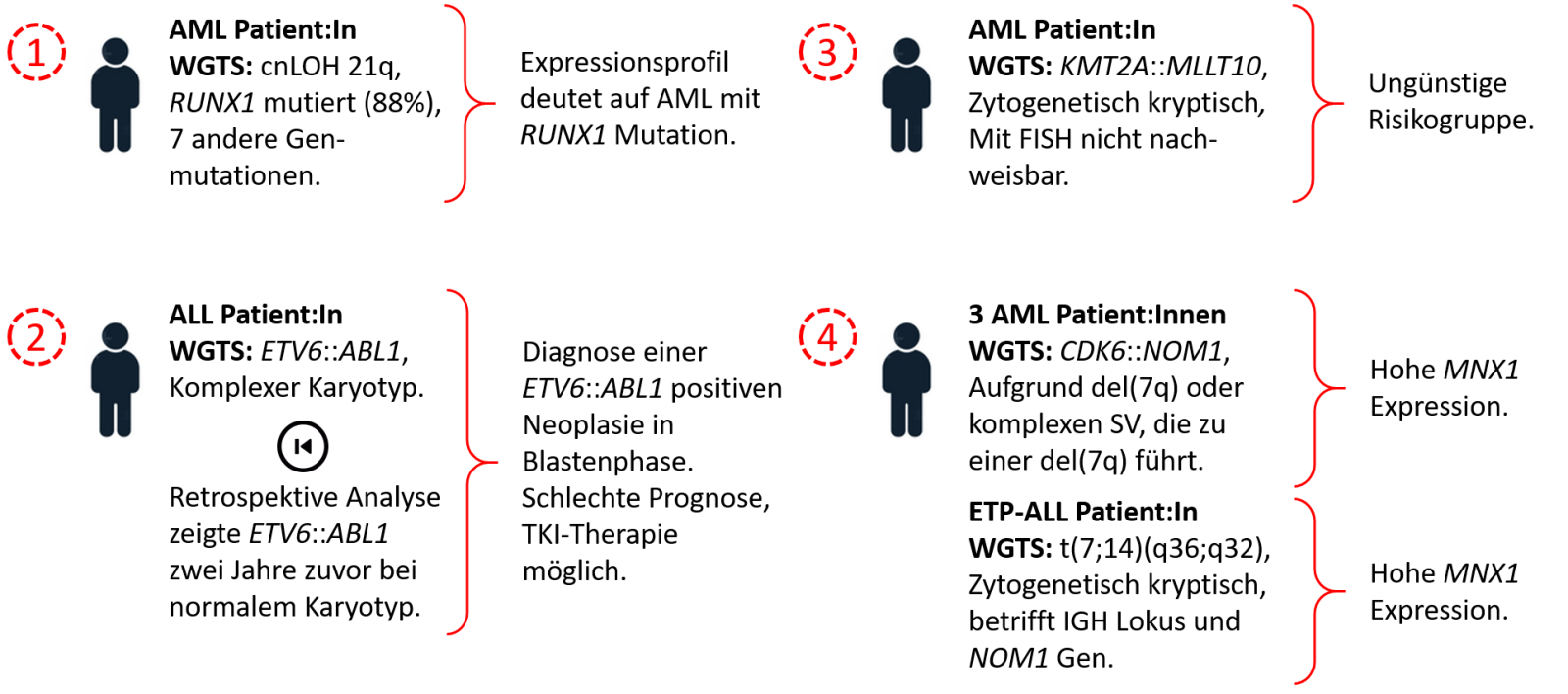
- Durch WGS konnte bei einem AML Patienten ein cnLOH 21q detektiert werden, das die hohe RUNX1 Mutationslast erklärte. Neben weiteren 7 Genmutationen zeigte auch das WTS-Expressionsprofil eine Zuordnung zu Patienten mit einer AML mit mutiertem RUNX1.
- WGS identifizierte auch eine zytogenetisch kryptische SV, die zu einem ETV6::ABL1-Rearrangement bei einem ALL-Patienten mit komplexem Karyotyp führte. Eine retrospektive Analyse zeigte, dass die Aberration bereits vorhanden war, als sich der Patient zwei Jahre zuvor mit einer MPN und einem normalen Karyotyp (CBA) vorstellte. Daher führte WGS zur Diagnose einer ETV6::ABL1-positiven Neoplasmasie in der Blastenphase (schlechte Prognose), das mit Tyrosinkinase-Inhibitoren behandelt werden könnte.
- In einem AML-Fall offenbarte WGTS ein KMT2A::MLLT10-Rearrangement, das zytogenetisch kryptisch und durch FISH nicht nachweisbar war (KMT2A-Break-Away-Sonde) und den Patienten der ungünstigen Risikogruppe zuordnete.
- Bemerkenswert ist, dass bei drei AML-Patienten eine 7q-Deletion oder eine komplexe strukturelle Veränderung, die zu einer 7q-Deletion führt, zu einem CDK6::NOM1-Fusionstranskript und einer hohen MNX1-Expression führte. Darüber hinaus ergab WGS bei einem Patienten mit ETP-ALL eine zytogenetisch kryptische Translokation t(7;14)(q36;q32), die den IGH-Locus und das NOM1-Gen betrifft und mit einer hohen MNX1-Expression einhergeht. Eine erhöhte MNX1-Expression als Folge von t(7;12)(q36;p13) ist ein sehr seltenes Ereignis bei pädiatrischer AML, aber unsere Ergebnisse könnten darauf hindeuten, dass die MNX1-Expression bei akuter Leukämie bei Erwachsenen eine wichtigere Rolle spielt als bisher bekannt.